
Table of Contents
Western blot technique is also known as the immunoblotting technique. Separation of mixture of proteins by electrophoretic techniques usually results in a complex pattern of protein bands or zones. Specific proteins are identified using this method.
The principle and the process of the Western Blot:
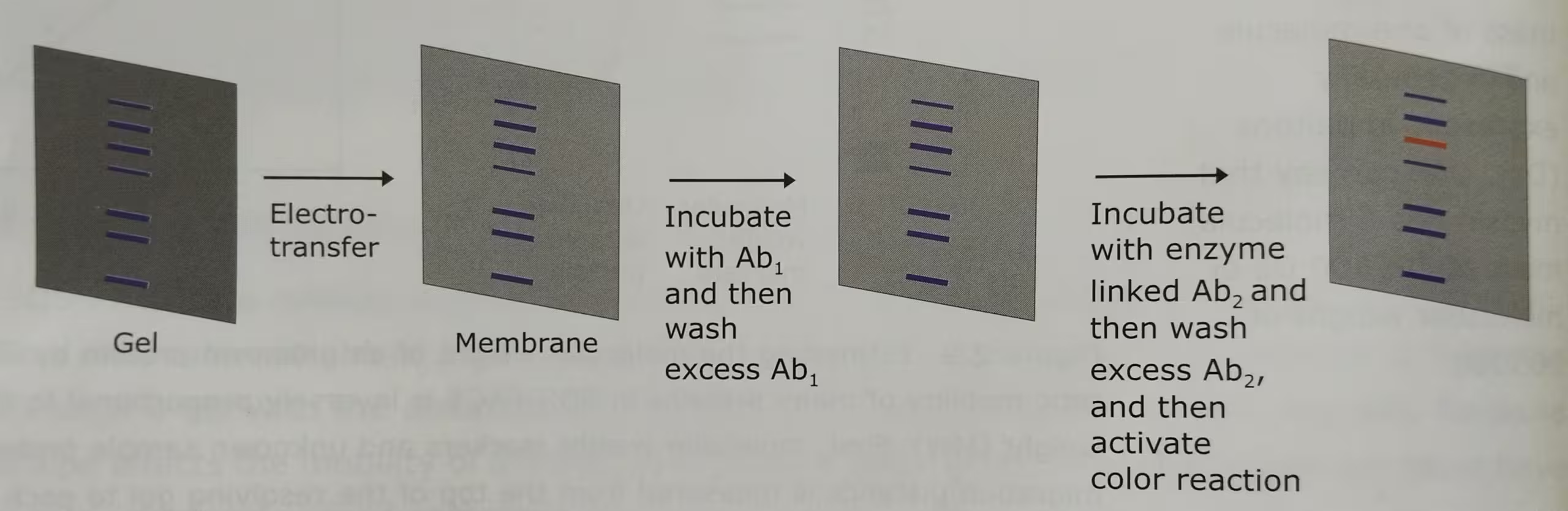
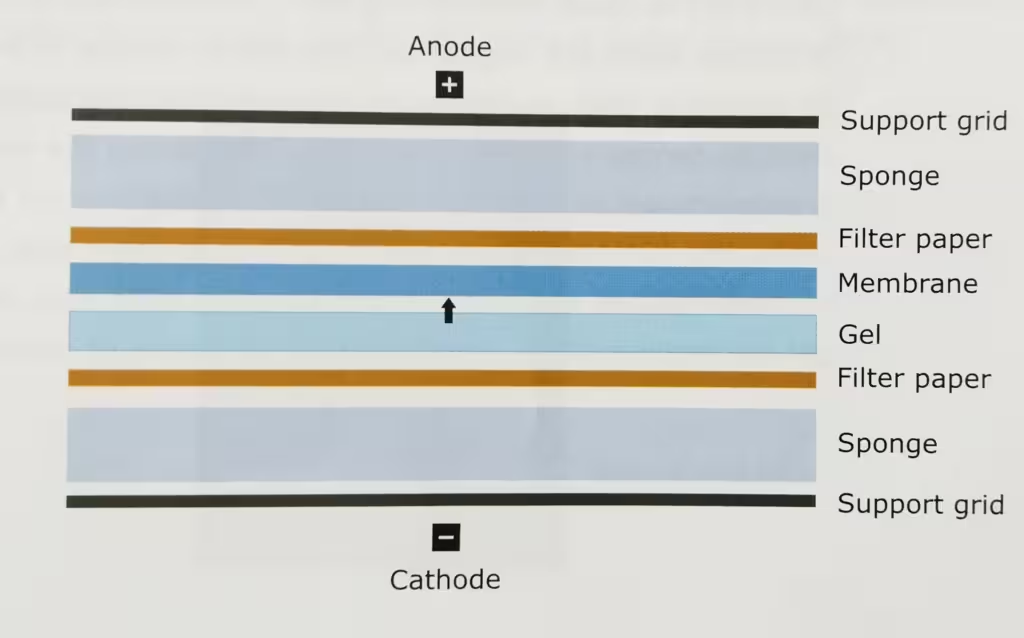
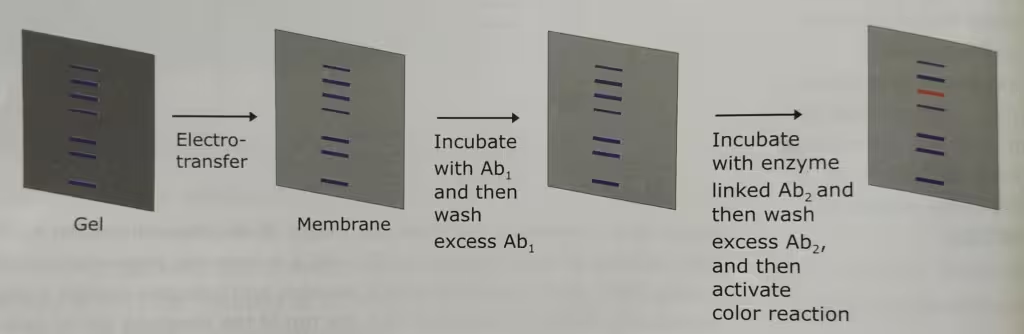
The technique Western Blot requires an antibody against the test protein. After the initial separation by an electrophoretic technique in a gel, the proteins are transferred (or blotted) from gel to a membrane, usually nitrocellulose or polyvinylidene difluoride (PVDF). Proteins are transferred from the gel to a membrane by applying an electric field perpendicular to the gel. During the transfer, the gel is at the negative electrode (cathode) side and membrane at the positive electrode (anode) side. Proteins that are coated with negatively charged SDS will move from the negative (anode) side, the gel to the positive side, the membrane.
The membrane that contains the resolved proteins will be first incubated with an unrelated protein to block nonspecific protein binding sites on the membrane, and then treated with a suitable primary antibody that is specific for the protein of interest.
Excess antibodies are then washed from the membrane and the bound antibody, which remains, is detected using a secondary antibody against the first.
The secondary antibodies are conjugated with the fluorescent or radioactive labels or enzymes that give a subsequent reaction with an applied reagent, leading to a coloring or emission of light, enabling detection.
The labelled secondary antibody which binds to the primary antibody is often used to enhance the sensitivity of the primary antibody- the antibody that reacts with the target molecule. In case of fluorescently labelled secondary antibodies, the amplification is usually several-fold.
Western blot protocol:
This is the general western blot protocol. This may vary laboratory to laboratory.
- Western blot protocol for protein estimation from HEK- 293 and MDA-MB-231 cells.
- Cell lysis: Thaw the PBS (Phosphate buffered saline) pelleted cell and discard the PBS Resuspend in 300 µl RIPA (Radio-Immuno Precipitation Assay) buffer and transfer the content into an Eppendorf (Incubate at 4°C for 30 minutes).
- Get sonication probe (for Eppendorf or MCT).
- Perform Bradford assay to determine the protein concentration in both pellet and supernatant (by taking duplets and BSA as control).
- Dissolve the pellet in RIPA buffer for making a suspension.
- Collect supernatant (contains proteins) in a separate MCT and do not discard the pellet.
- Centrifuge at 12,000 rpm for 10 minutes (to pellet down cell membrane and other debris).
- Sonicate to lyse the cells (8 cycles for 10 seconds with breaks of 10 seconds).
- Prepare SDS PAGE gel (12% Resolving and 5% stacking) and samples for loading along with protein ladder.
- Prepare the set-up for SDS-PAGE by putting gel into the cassettes and then to the chamber.
- Load the gel cassettes and SDS running buffer (always remove the combs inside the buffer) once the comb is removed carefully load the samples (25 µl) and protein ladder (2-3µl) into wells and run the gel (90V).
- Protocol for transfer to PVDF
- Track the dye till bottom. Prepare for membrane transfer.
- Take three boxes for (1). Distilled water: to keep the gel (2) Methanol: for activation of the PVDF membrane (3) Transfer Buffer: to keep the sponges and filter papers wet.
- Soak the gel in distilled water remove the gel from the glass plates carefully, PVDF membrane in methanol for 5 minutes and sponges with filter paper in transfer buffer.
- Set the cassettes properly and keep it inside the transfer chamber (red and black side should be kept properly), add flee and load the transfer buffer inside the transfer tank.
- Keep the set up at 4°C on stirrer for 1-2 hours, at 29-50V.
- heck the PVDF membrane and once the transfer is done take out the transfer chamber from 4°C.
- Discard the gel and wash the PVDF membrane in RO water.
- Cut the extra gel and kept the gel according to the molecular weight of the protein. Cut the PVDF membrane according to the size of the gel.
- Set the cassettes by making a sandwich: black cassettes (cathode ’-‘)- sponge- 4 filter paper- gel-PVDF-4 filter paper-sponge-red cassettes (anode ’+’) and remove air bubbles with the help of glass rod when putting filter paper.
- [Note: Keep everything moist with transfer buffer]
- Wash the membrane with chilled 1X PBST buffer (Three times 3 minutes each on shaker at room temperature).
- After washing, add blocking solution: 3% BSA for 60 minutes kept on shaker with slow speed.
- Second wash with Chilled 1X PBST (three times for 3 minutes each at room temperature) on shaker.
- Incubate with primary antibody (1:2000) (at 4°C overnight on slow shaker).
- Retrieve primary antibody.
- Wash with Chilled 1X PBST (three times 3 minutes each at room temperature with on shaker)
- Incubate in Secondary antibody (1:750) at room temperature for 2 hours on shaker.
- Retrieve the secondary antibody and wash with Chilled PBST three to four times 3 minutes each at room temperature.
- Keep it in RO water till blot development.
- Prepare for blot development.
- Prepare ECL mix (1:1; Luminol (0.5 ml): hydrogen peroxide (0.5 ml) in a separate falcon tube and cover it with foil wrap).
- Discard water, Add ECL mix to PVDF membrane blot and mix gently for 3-4 minutes and
- then check for chemiluminescence (cover the box with foil to avoid light exposure) on Image quant application.
Other related links:
Cell reviving process: https://thebiologyislove.com/the-basic-steps-of-the-cell-reviving-process/